


{kind=link}
Epidemiologia delle malattie delle mani e dei piedi (HFMD) nel Guangdong, Cina, 2013-2021
La segnalazione delle malattie delle mani e dei piedi (HFMD), la raccolta dei campioni e il sistema di tipizzazione molecolare sono illustrati nella Figura 1. Secondo il sistema provinciale di segnalazione delle malattie notificabili, sono stati segnalati in totale 3.009.233 casi clinicamente confermati di malattie delle mani e dei piedi (HFMD). Guangdong, Cina, 2013-2021, con il maggior numero di casi segnalati nel 2014 (N= 429.617) e il più basso nel 2020 (N= 60501) (Figura 2a). Il test RT-PCR per EV e i genotipi specifici EV-A71 e CVA16 era richiesto per i campioni sospetti raccolti da HFMD nel 2013-2016, che si sono estesi a EV-A71, CVA16 e CVA6 nel 2017. Sono stati analizzati in totale 36.461 campioni sospetti da HFMD raccolti per la profilazione. Test genetici nel periodo 2013-2021, di cui 26.086 (71,54%, 26.086/36.461) sono risultati positivi all’HIV. Per la distribuzione del genotipo EV, la proporzione di EV-A71 e CVA16 ha oscillato nel tempo. È stato osservato che l’epidemia di CVA16 aumenta ogni due anni con un’incidenza relativamente più elevata (>25%) rilevata rispettivamente nel 2014, 2016, 2018 e 2021 (Figura 2a). Al contrario, il tasso di positività è diminuito (
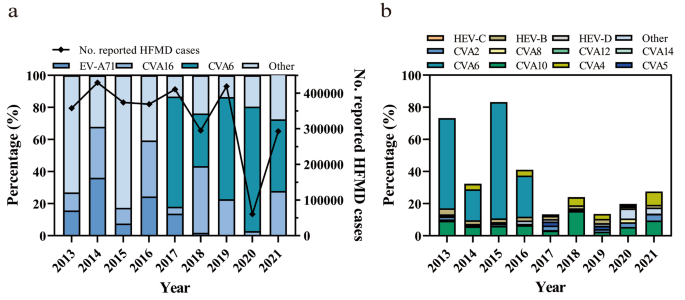
Distribuzione di enterovirus nei casi di HFMD nel Guangdong, Cina, 2013-2021. (UN) Distribuzione di EVA71 e CVA16 nel 2013-2016 e distribuzione dei genotipi EVA71, CVA16 e CVA6 nel 2017-2021; (B) Distribuzioni del genotipo di enterovirus in altri casi di HFMD positivi a enterovirus (HFMD non EVA71 e non CVA16 nel 2013-2016; e HFMD non EVA71, non CVA16 e non CVA6 nel 2017-2021). EV, enterovirus. CV, coxsackievirus. Malattia delle mani, dei piedi e della bocca (HFMD).
Diversamente dal modello eziologico della malattia delle mani e dei piedi (HFMD) osservato nel 2008-2013, dal 2013 è stato osservato un aumento nel rilevamento di genotipi EV diversi da EV-A71 e CVA16. I nostri precedenti rapporti hanno evidenziato la dominanza di CVA6 nei casi di HFMD (HFMD) nel Guangdong entro la fine del 2013. [20, 27]. Il continuo aumento dell’incidenza del CVA6 dal 2013 al 2021 suggerisce che questo genotipo sta diventando endemico nella regione. Dei 3.119 campioni identificati come EV non EV-A71/CVA16/CVA6, CVA10 ha rappresentato la percentuale maggiore tra il 2013 e il 2021, con un picco nel 2018 con una prevalenza del 64,60% (500/774). In particolare, CVA4 ha mostrato una significativa tendenza al rialzo, raggiungendo il tasso di rilevamento più alto nel 2021 al 29,82% (243/815). Inoltre, CVA2, CVA5 e CVA8 sono stati comunemente rilevati in casi sospetti di HFMD, suggerendo un modello eziologico multiforme di HFMD nel Guangdong, in Cina (Figura 2 B).
La diffusione del CVA10 dal 2013 al 2021 ha spinto a indagare sulle caratteristiche epidemiologiche dell’infezione da CVA10. Abbiamo eseguito un’analisi retrospettiva di 614 casi di infezioni da CVA10 e 467 casi di infezioni da EV-A71 segnalati al sistema provinciale di sorveglianza delle malattie notificate tra il 2017 e il 2021. La stragrande maggioranza, il 98,37% (604/614) delle infezioni da CVA10 e il 96,36% (450/467) delle infezioni da EV-A71, si è verificata in bambini di età inferiore a 5 anni. In particolare, i tassi di infezione più elevati sia per le infezioni CVA10 che per quelle EV-A71 sono stati osservati nel gruppo di bambini di età compresa tra 1 e 2 anni. L’età media dei pazienti con CVA10 era di 1,8 anni, che è significativamente inferiore all’età media di 2,3 anni per l’infezione da EV-A71.SP > 0,05), come mostrato nella Figura 3.
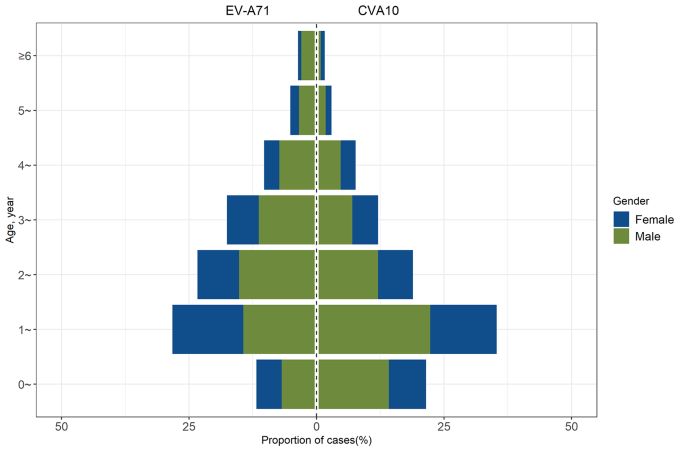
Distribuzione per età e sesso dei casi EVA71 e CVA10 con HFMD dal 2017 al 2021
Struttura filogenetica del gene VP1 e del genoma virale
Ad oggi, nel database pubblico è presente solo un numero limitato di sequenze del genoma, nonché di sequenze del gene VP1, per CVA10. Al fine di ottenere informazioni dettagliate sulla distribuzione evolutiva dei virus e delle mutazioni potenzialmente associate alla crescente epidemia, nel nostro studio abbiamo generato un totale di 78 sequenze complete o quasi complete del genoma virale CVA10, che rappresentano campioni raccolti dal 2010 al 2021. è stato eseguito includendo 402 sequenze del genoma CVA10 disponibili nel database pubblico.
Innanzitutto, tutte le sequenze del genoma CVA10 sono state allineate a AY421767 come genoma di riferimento. Il corrispondente gene VP1 (2477-3377nt) è stato poi sottoposto a splicing e utilizzato nel costrutto filogenetico. Abbiamo seguito lo schema di classificazione VP1 di studi precedenti [28, 29]Tutte le sequenze possono essere classificate in sei gruppi di geni. Le somiglianze genetiche VP1 tra i diversi gruppi di genotipi variavano dal 74,94 all’86,47%, con i genotipi dei gruppi A e D che mostravano la maggiore divergenza (Tabella 1). Nel ceppo è stata osservata una circolazione endemica, con tutte le sequenze genomiche del Guangdong CVA10 appartenenti al genogruppo C. Secondo la sequenza genomica, la crescente prevalenza di CVA10 è stata molto probabilmente causata dai due sottogruppi di virus del genogruppo C che rappresentano il genogruppo più grande dopo il 2015 (Figura 4a, pannello superiore).
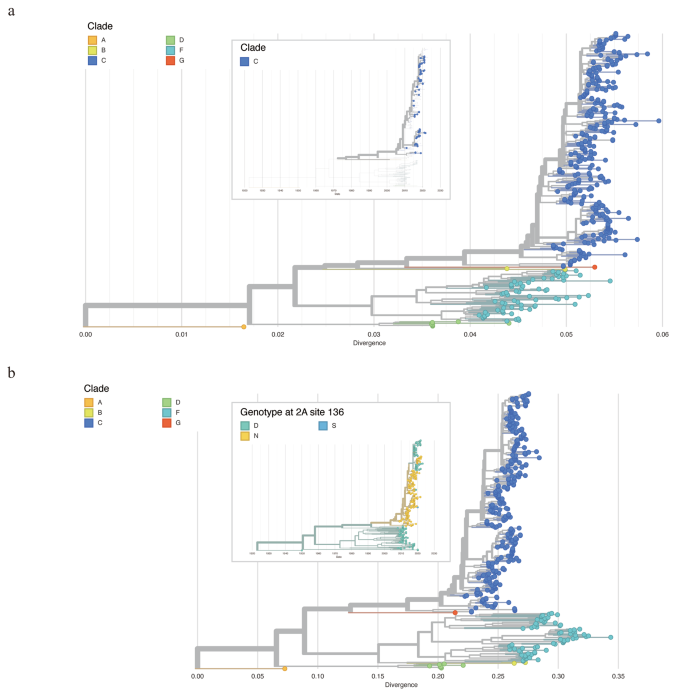
Albero di massima verosimiglianza di CVA10 costruito sulla base di VP1 (a) e del genoma completo (b). Miniatura nel pannello superiore (UN) ha mostrato l’albero filogenetico su scala temporale di VP1 e vengono mostrate solo le sequenze del Guangdong generate in questo studio. Miniatura nel pannello superiore (B) mostra lo sviluppo evolutivo del genoma CVA10. L’albero è colorato dall’amminoacido in posizione 136 della proteina 2
In secondo luogo, abbiamo ricostruito la filogenesi utilizzando la sequenza del genoma CVA10 e il clade è stato annotato in base ai risultati della classificazione VP1 (Figura 4b). La ricostruzione della sequenza ancestrale utilizzando il metodo della massima verosimiglianza ha indicato le probabili sequenze ancestrali per ciascun nodo e le corrispondenti mutazioni di amminoacidi che si verificano in ciascun ramo. È interessante notare che la ricostruzione della sequenza ancestrale ha rivelato il ramo interno che ha dato origine al sottogruppo C appena emergente, che comprende la maggior parte delle sequenze CVA10 raccolte dopo il 2017 (84 su 202, 41,58%) e che contiene un amminoacido unico, N136D. , nella proteina 2a. Questa mutazione non sinonimo è in netto contrasto con le 104 mutazioni sinonime identificate nell’intera sequenza del genoma. (Figura 4b).
In particolare, l’amminoacido in posizione 136 è stato ripetutamente modificato durante l’evoluzione del virus. Il ramo interno che porta al genogruppo C di CVA10, comprese le mutazioni N136D e le sequenze di tutti gli altri genogruppi, era un acido aspartico (Asp, D) in posizione 136 della proteina 2 A. La mutazione inversa può essere osservata nel nuovo sottogruppo che emerge dal gruppo C As menzionato sopra. Inoltre, abbiamo osservato che un piccolo gruppo nella coorte emergente aveva la mutazione N136D nella proteina 2A, accompagnata da altre mutazioni di aminoacidi nelle proteine 3A, 3C e 3D. La funzione biologica delle mutazioni nella posizione 136 della proteina 2A deve essere ulteriormente analizzata per comprendere il meccanismo dell’evoluzione del virus CVA10.
Frequente ricombinazione tra CVA10 e altri enterovirus
Le annotazioni dei cluster genetici sulla sequenza VP1 e sull’intera sequenza del genoma di CVA10 hanno rivelato la differenza nella struttura evolutiva di questi due (Figura 4a-b). In dettaglio, il genogruppo B della sequenza CVA10 era strettamente correlato al genogruppo G e al genogruppo C nella filogenesi VP1 ma era raggruppato con ceppi del genogruppo F nelle sequenze della sequenza del genoma. Questa discrepanza ha evidenziato la possibilità di ricombinazione all’interno del pool genetico o di ricombinazione tra specie EV durante l’evoluzione del virus CVA10. Per indagare possibili indizi sulla ricombinazione genetica, abbiamo sequenziato rispettivamente le regioni P1, P2 e P3 del genoma CVA10. Allo stesso modo, il cluster genetico G di CVA10 era strettamente correlato al cluster genetico F nella sequenza di sequenze codificanti proteine non strutturali (P2 e P3) che era incoerente con la filogenesi della regione codificante proteine strutturali (P1) (Figura 1a-c supplementare) . È noto che la ricombinazione è comune tra le specie EV nelle regioni codificanti proteine non strutturali [30, 31]. Per chiarire la relazione tra CVA10 e altri tipi di EV, abbiamo ricostruito le sequenze di P2 e P3 includendo tutte le sequenze strettamente correlate di altri EV. Per la regione P2, tutti i cluster di geni CVA10 sono stati dispersi in tutto l’albero filogenetico e raggruppati con altre specie EV piuttosto che con altre sequenze di cluster di geni CVA10, evidenziando il potenziale di ricombinazione interspecie (Figura 5a). Nello specifico, i frammenti P2 del genogruppo F rientravano in tre diversi gruppi principali, indicando una possibile ricombinazione tra CVA10 genogruppo F, CVA2, CVA4, CVA8, EVA114, EVA120 e altri tipi. Il genogruppo C, i ceppi CVA10 predominanti nella Cina continentale, erano raggruppati insieme, con una sola sequenza (MF422532) che forse presentava ricombinazione con i tipi EV-A71 e CVA2. Risultati simili sono stati trovati per la regione P3 (Figura 5B), dove le sequenze di cluster di geni F erano strettamente correlate ad altri tipi di EV e raggruppate in più gruppi. Le complesse interazioni tra il cluster genetico F di CVA10 e altri EV nelle sequenze codificanti proteine non strutturali hanno indicato il potenziale per una circolazione diffusa di questo cluster genetico nella popolazione. Per confermare la presenza di eventi di ricombinazione nel cluster genetico F di CVA10, sono state eseguite analisi Simplot con altri EV (Figura 6). I risultati hanno rivelato eventi multipli di ricombinazione tra il gruppo genetico F di CVA10, EVA120, CVA2 e CVA4 nelle regioni codificanti P2 con valori di bootstrap superiori al 70%.
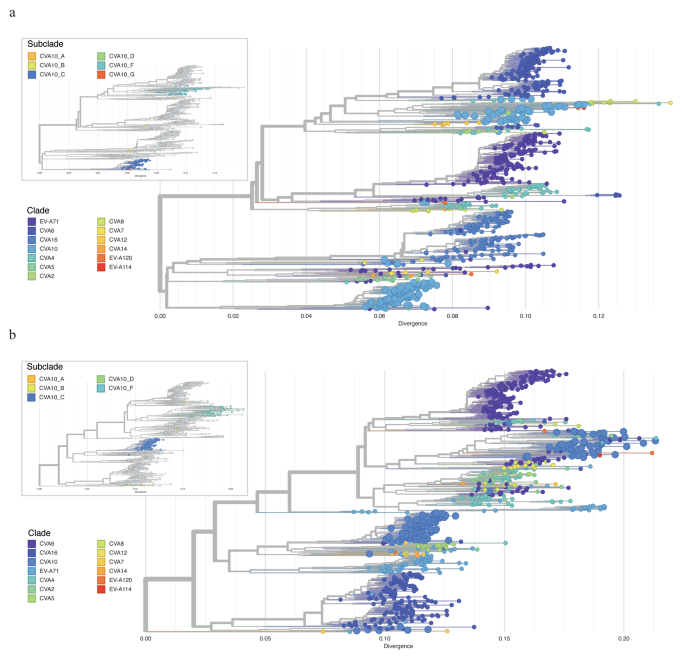
Gli alberi di massima verosimiglianza sono stati ricostruiti includendo P2 (UN) e P3 (B) Regione CVA10 e altre sequenze di enterovirus strettamente correlate. Le micropiastre hanno dimostrato la posizione filogenetica dei sottolignaggi CVA10

Analisi di ricombinazione del cluster genico F di CVA10. Analisi di ricombinazione della scansione bootstrap tra il genogruppo F di CVA10 ed EVA120 (UN),CVA2(BCVA4(C). I ceppi rappresentativi (EVA120: MT081367, CVA2: MF422537, CVA4: OM417121) sono stati utilizzati rispettivamente come sequenze di query. La linea tratteggiata indica il 70% di supporto bootstrap. La struttura del genoma di CVA10 è stata annotata secondo il ceppo di riferimento CVA10 AY421767.

Elena Ferrante scrive per SanSevero.TV occupandosi di attualità, politica, economia, tecnologia, sport, intrattenimento e lifestyle. Si dedica a offrire informazioni chiare e utili, raccontando eventi e temi di interesse con un approccio accessibile, equilibrato e orientato ai lettori.